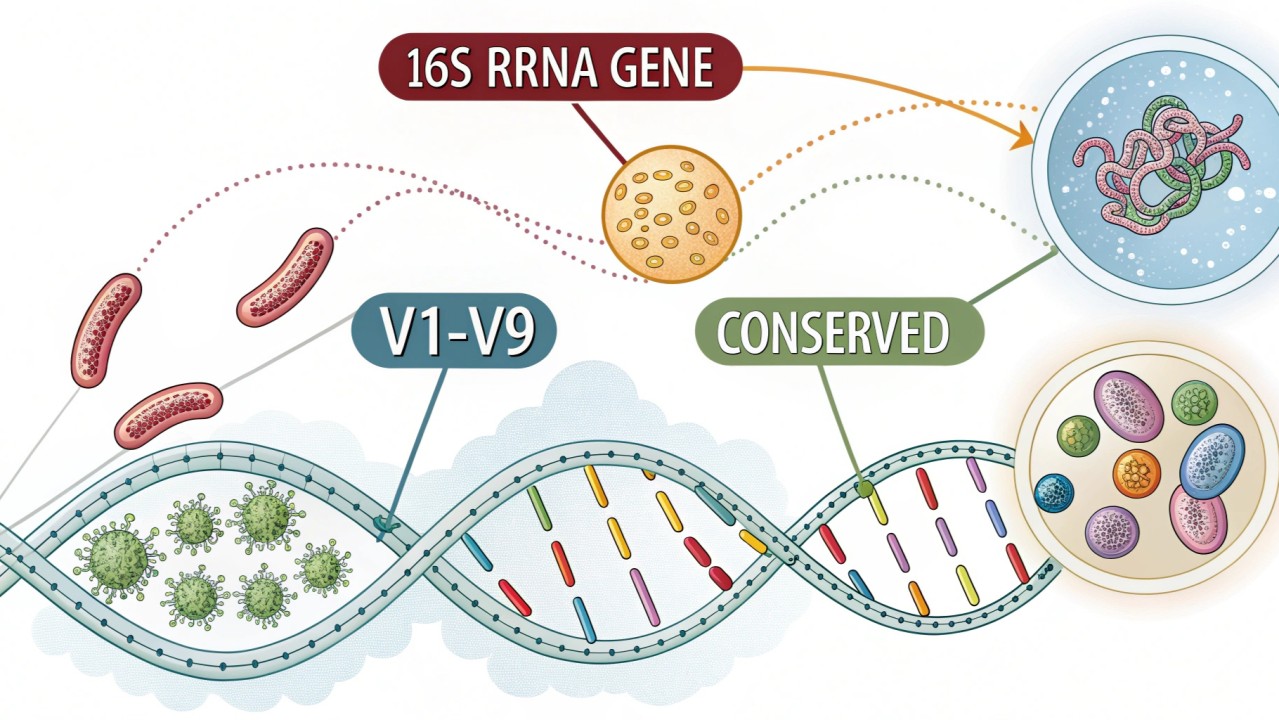
The 16S ribosomal RNA (rRNA) gene is a component of the small subunit of the prokaryotic ribosome and is universally present across bacteria, making it an essential molecular marker in microbiology. Its structural stability and the presence of both highly conserved and variable regions allow researchers to identify and classify bacteria accurately. The 16S rRNA gene’s relevance to microbiome studies lies in its ability to provide detailed insights into microbial diversity, population dynamics, and community structure across diverse environments, ranging from human health to agriculture and environmental ecosystems.
The 16S rRNA gene comprises nine distinct hypervariable regions (V1-V9), interspersed among conserved segments, providing phylogenetic resolution at various taxonomic levels. This structural characteristic makes 16S rRNA an ideal target for microbial community profiling and taxonomic identification through high-throughput sequencing methods, particularly next-generation sequencing (NGS).
Different hypervariable regions possess varying taxonomic resolution capabilities, influencing their suitability for specific microbiome studies. For instance, regions V1-V3 offer robust discrimination at the genus and species level within specific microbial communities, often utilized in gut microbiome research. The V3-V4 regions are highly favored due to their balanced length, offering excellent species-level resolution across diverse microbial taxa, making them ideal for comprehensive community analyses in environmental and clinical samples.
Conversely, V4 alone, although shorter, is frequently chosen for large-scale microbiome studies due to its consistent and reproducible performance across diverse microbial taxa, coupled with efficient sequencing workflows, notably demonstrated in extensive human microbiome projects. The V5-V6 regions provide valuable phylogenetic information, often utilized when studying environmental samples or complex communities requiring deeper taxonomic resolution, especially within the Firmicutes and Actinobacteria phyla.
Next-generation sequencing (NGS) technologies amplify these hypervariable regions through PCR-based amplicon sequencing, utilizing universal primers designed specifically to target these regions. The resultant amplicons are then sequenced on high-throughput platforms such as Illumina MiSeq or NovaSeq, allowing simultaneous analysis of thousands of samples with unprecedented accuracy and speed. Bioinformatics pipelines subsequently process the sequencing data, aligning the sequences against curated databases (e.g., SILVA, Greengenes, or RDP), assigning taxonomy with high confidence based on sequence similarity.
Historically, microbial communities were characterized using operational taxonomic units (OTUs), defined typically by clustering sequences at a 97% similarity threshold. OTU-based analyses provided a practical framework for community comparisons but were limited by clustering-induced inaccuracies and a lack of precision at finer taxonomic levels. In contrast, modern NGS approaches have shifted towards Amplicon Sequence Variants (ASVs), exact sequences obtained through advanced bioinformatics tools such as DADA2, Deblur, or UNOISE. ASVs offer higher resolution, reproducibility, and comparability across studies, enabling precise differentiation of closely related taxa and significantly improving the accuracy of microbial community profiling.
This targeted approach allows researchers to effectively characterize microbial diversity and abundance at various taxonomic ranks—from phylum to species—facilitating a detailed understanding of microbial community composition, dynamics, and functions. As a result, NGS-driven 16S rRNA amplicon sequencing using ASVs has become instrumental in advancing microbiome research across multiple domains, from clinical diagnostics and therapeutics to environmental monitoring and biotechnology.