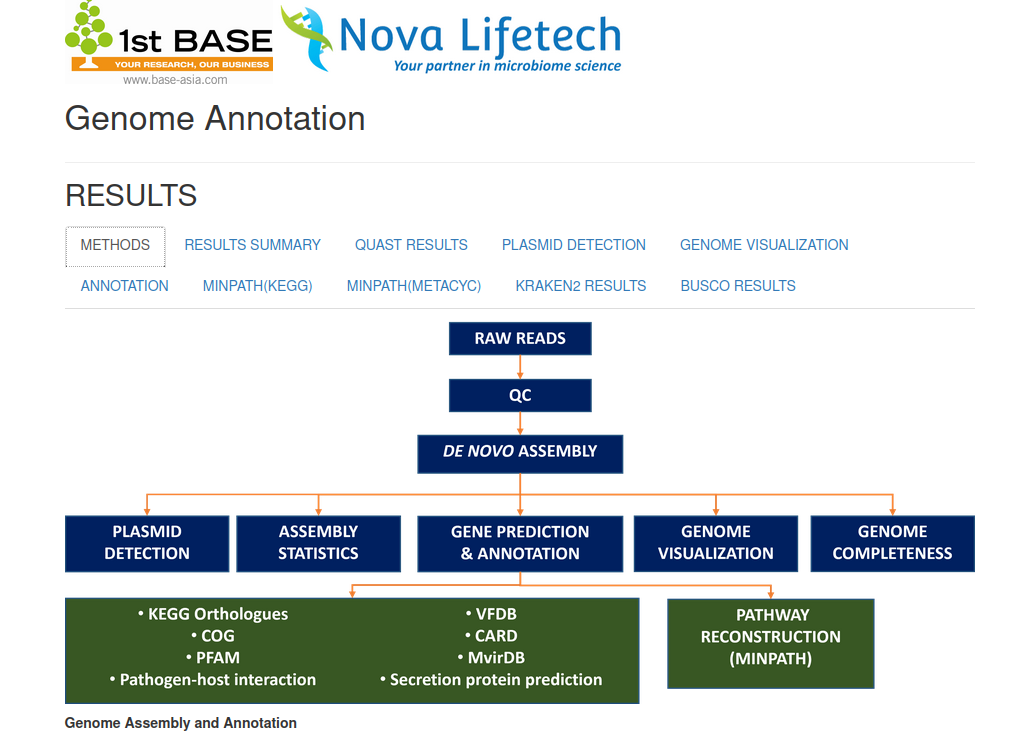
WGS Bioinformatics Services Demo
This WGS demo showcases a customizable bioinformatics services pipeline designed for bacterial genomes. It includes de novo assembly, plasmid detection, gene prediction, functional annotation using databases like KEGG, VFDB, and CARD, and pathway reconstruction via MinPath. The workflow also supports genome visualization, completeness assessment, and analysis of pathogen–host interactions. For eukaryotic genomes, the pipeline can be adapted to handle complex gene structures and larger genome sizes. For viral genomes, reference-guided assembly and virus-specific annotation tools are used. This flexible system supports bacterial, viral, and eukaryotic genome analysis across a variety of research applications.
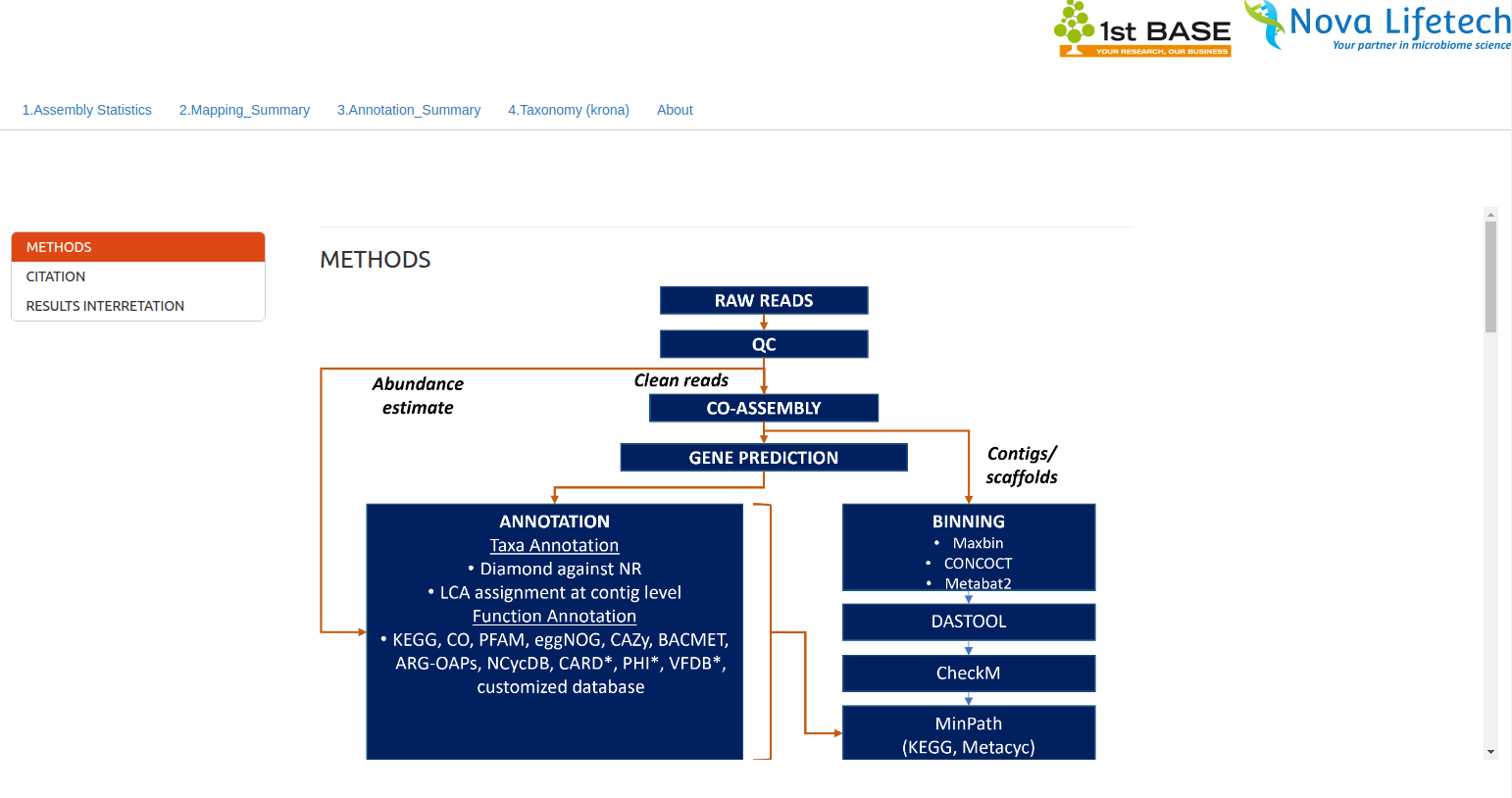
Metagenomics Bioinformatics Services Demo
This metagenomics demo showcases flexible bioinformatics services for diverse sample types and research questions. Starting from raw paired-end reads, the pipeline performs quality control, co-assembly, and gene prediction, followed by taxonomic and functional annotation using DIAMOND and databases like KEGG, eggNOG, CAZy, and CARD. Binning and MAG recovery are handled via tools such as MetaBAT2, MaxBin, and CONCOCT, with quality checks using CheckM. The workflow is highly customizable, allowing users to adjust binning strategies, annotation sources, and statistical methods (e.g., DESeq2, LEfSe, PERMANOVA). Results can be visualized through diversity plots and functional profiles, offering a scalable solution for exploratory and hypothesis-driven research.
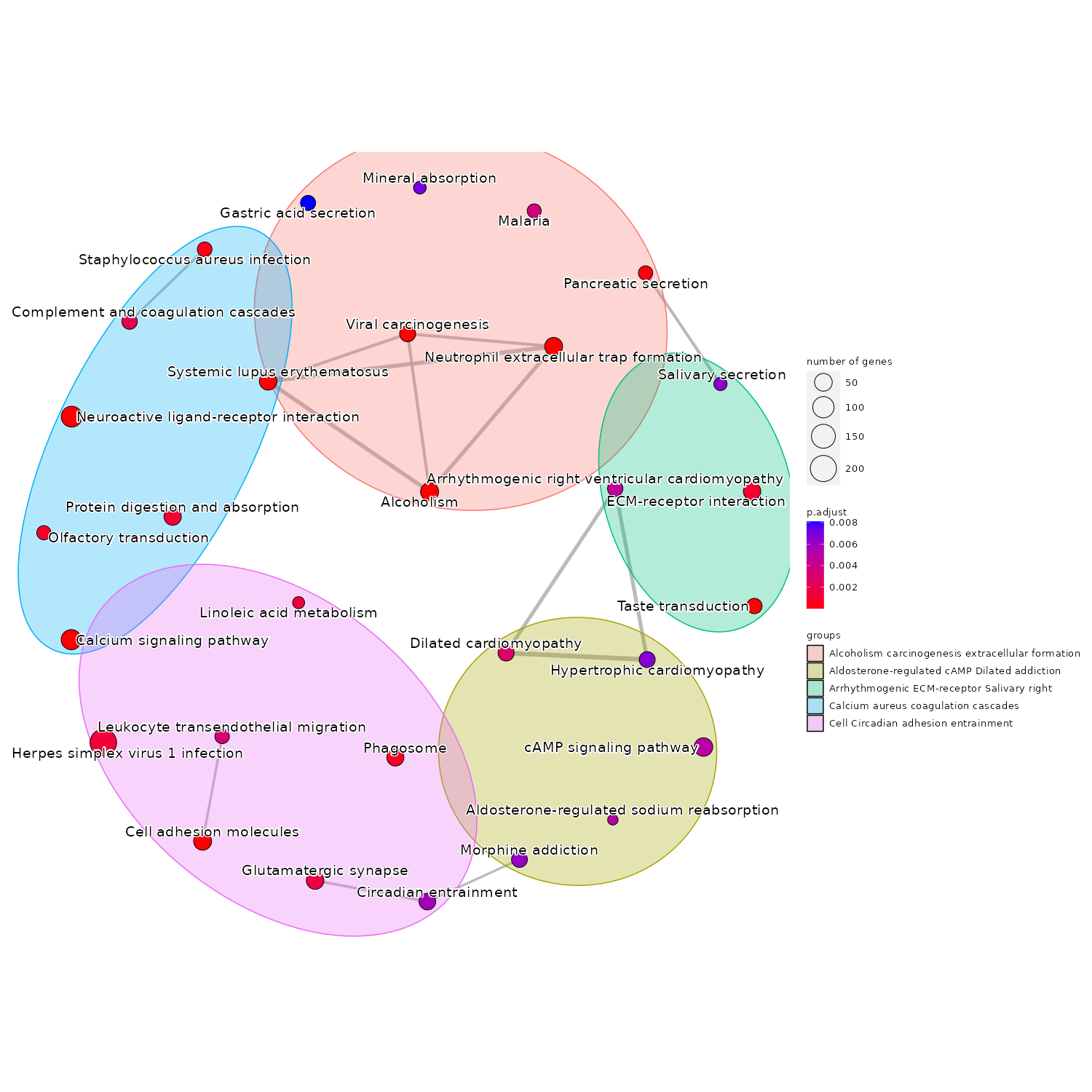
RNA-seq Bioinformatics Demo Report
This RNA-seq demo showcases flexible bioinformatics services for transcriptomic analysis across a range of organisms. Using user-defined reference genomes, the workflow begins with raw RNA-seq data and includes alignment via STAR or Bowtie2, read quantification with featureCounts, and differential expression analysis using DESeq2 or edgeR. Downstream analyses include PCA, heatmaps, volcano plots, gene enrichment, pathway analysis, and fusion gene detection (for human data). The pipeline is highly adaptable, making it suitable for diverse experimental designs and biological questions.

Variant Calling Bioinformatics Demo
This variant calling demo report presents a flexible pipeline using FreeBayes v1.3.6, with customizable components based on project needs. Raw sequencing reads are aligned to a reference genome using tools such as BWA (for Illumina) or Minimap2 (for PacBio/Nanopore), followed by duplicate marking and sorting via Picard. Variant calling is performed with FreeBayes, applying filters such as minimum coverage (<10). Results are visualized using IGV or DISCVRSeq. The pipeline is modular and can be adapted with alternative open-source tools (e.g., GATK, SAMtools, DeepVariant) to suit different organisms, data types, and analytical objectives.